
-
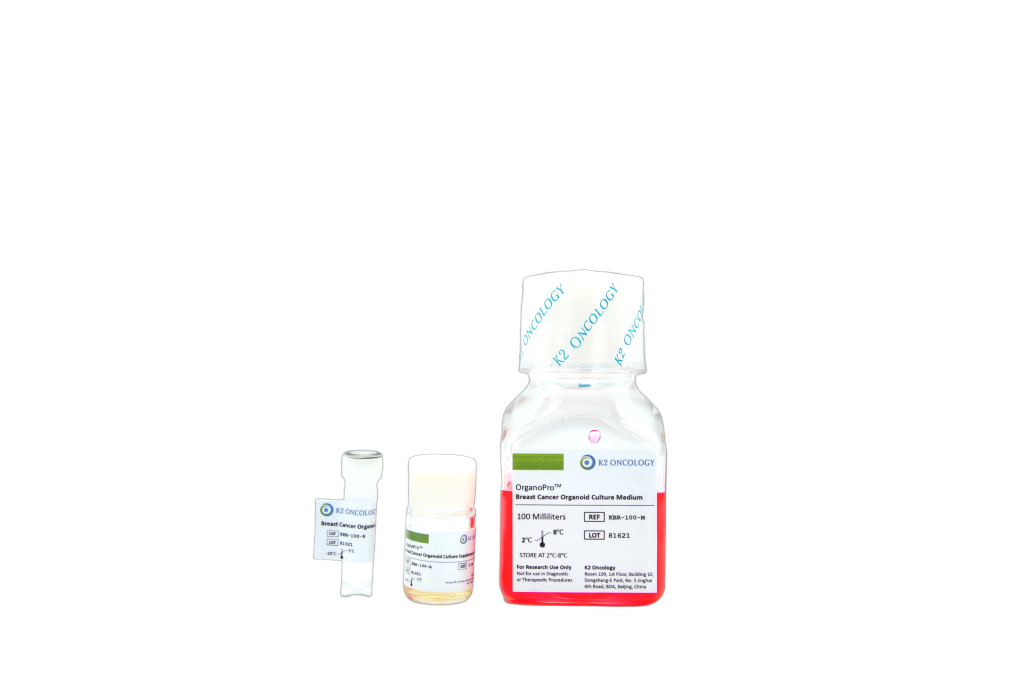
乳腺癌类器官培养试剂盒
KBR-100
-

乳腺癌类器官培养试剂盒
KBR-1000

OrganoPro™人源乳腺癌类器官培养基套装
KBR-100 包含以下产品
- OrganoPro™乳腺癌类器官培养基100mL
- OrganoPro™乳腺癌类器官添加剂成分A 2mL
- OrganoPro™乳腺癌类器官添加剂成分B 1mL
KBR-1000 包含以下产品
- OrganoPro™乳腺癌类器官培养基 1000mL
- OrganoPro™乳腺癌类器官添加剂成分A 10mL x 2
- OrganoPro™乳腺癌类器官添加剂成分B 10mL
科途医学 科学家

名字
PhD
我们的科学家向您推荐
我们的产品简化了实验流程,集成多种因子,无需单独优化,扩增潜力高,14天内细胞数量可达到1×10^6。适用于多种培养形式,包括基质胶、低吸附孔板和生物反应器悬浮培养。GMP级别生产条件下制备,批次质量稳定,试剂含量是常规市售干细胞培养基的2倍,实现极佳的成本效益比。让复杂的培养变得简单快速,让科研变得更高效。
概览
此产品基于 Simumatrix 技术平台,通过工业化高通量筛选,针对中国高发的肿瘤类型进行培养基优化筛选而开发出的类器官培养基产品,可用于乳腺癌的类器官培养。
产品优势/特点:
- 简单易用,节省验证时间:提供详细操作方案,产品使用简便,节省研究者大量类器官培养摸索验证时间;
- 肿瘤组织覆盖类型广:覆盖多达15个组织瘤种,>900种肿瘤驱动基因突变模型;
- 扩增潜力高:自研高活力高稳定性WNT与RSPOs,支持肿瘤类器官多代次连续稳定培养;
- 肿瘤类器官验证数据齐备:多维类器官验证数据的整合,类器官驱动基因突变及表达谱,类器官组织病理学验证及类器官药敏数据等。
- 多篇高分文献应用:多篇高分文献应用,口碑卓越;
- 自主研发,产能充足,性价比高:全自研生产,源头品控,产品性价比高。
产品组成:
| 产品名称 | 货号 | 规格 | 储存温度 | 保质期 |
|---|---|---|---|---|
| OrganoPro™ Breast Cancer Organoid Culture Medium 人源乳 腺癌类器官基础培养基 | KBR-100/1000-M | 100mL / 1000mL | 2-8°C | 12个月 |
| OrganoPro™ Breast Cancer Organoid Culture Supplement A (50X) 人源乳腺癌类器官培养基添加剂 A (50X) | KBR-100/1000-A | 2mL / 20mL | -20°C | 12个月 |
| OrganoPro™ Breast Cancer Organoid Culture Supplement B (100X) 人源乳腺癌类器官培养基添加剂 B (100X) | KBR-100/1000-B | 1mL / 10mL | -20°C | 12个月 |
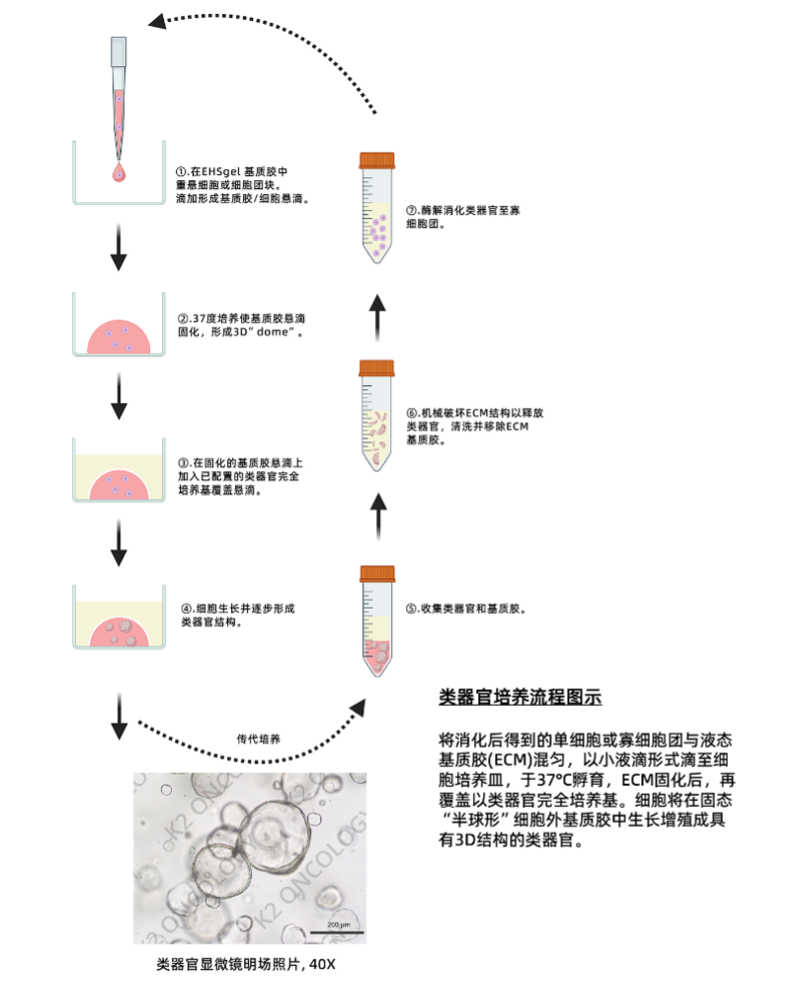
类型
类器官培养基
适用细胞
乳腺癌类器官
物种
人类
应用
乳腺癌类器官
商标
OrganoPro™
产品使用说明及支持信息
在产品文档中查找支持信息和使用说明,或在下方探索更多
| 文档类型 | 产品名称 | Catalog # |
|---|---|---|
| User manual | OrganoPro™乳腺癌类器官培养基套装 | KBR-100 KBR-1000 |
资源及文献引用
相关资源及文献引用
COA查询
根据货号和批次号,在线查询已购买产品的COA证书
请输入产品货号(REF or CAT Number) 和产品批号(LOT Number) ,查询COA证书文件。
产品货号和产品批号均显示在产品标签上对应位置(如右侧示意图所示)
产品货号和产品批号均显示在产品标签上对应位置(如右侧示意图所示)
产品货号
﹡
产品批号
﹡
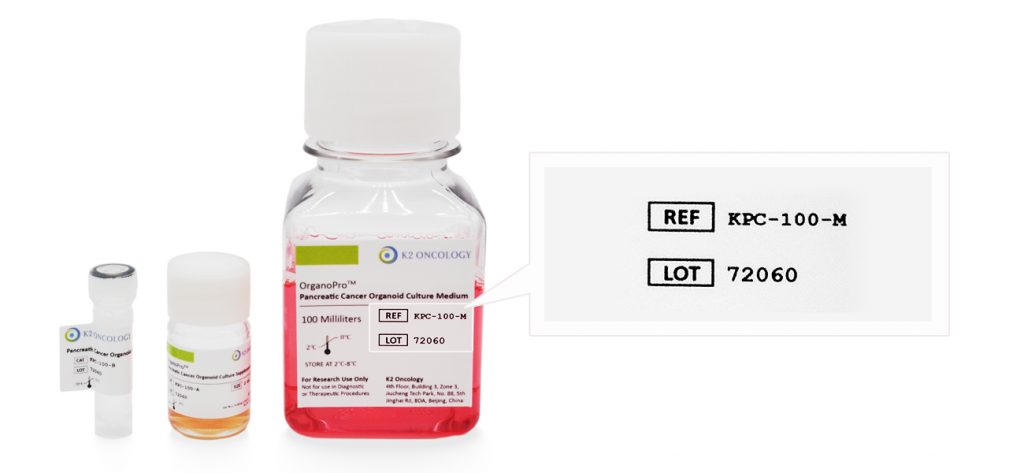
Ref/Cat#
产品货号,每个产品对应的独立货号
Lot#
批次编号,同一产品不同批次会有不同批号,请您在产品包装找到对应批号
